Predicting molecular structures from experimental data is a fundamental challenge in molecular sciences, crucial for applications such as drug discovery, materials science, and sustainable energy solutions. Traditional structure prediction methods are both computationally expensive and time-intensive. Advances in artificial intelligence (AI) and machine learning (ML) now offer an alternative, enabling more efficient and accurate structure predictions based on experimental and computational data.
Our journey towards an AI-accelerated pipeline for structure prediction began with developing machine learning methods to explore the high-dimensional space of molecular configurations and identify energy barriers along potential reaction paths.
Machine learning approach for energy barrier estimation
Energy barriers determine different molecular con-formations and are crucial for understanding molecular properties. We developed an ML framework to estimate reaction energy barriers without explicit and expensive transition state calculations. Using a dataset of over 11,000 reactions, this approach employs kernel ridge regression (KRR) to predict energy barriers based on molecular descriptors, including Coulomb matrices, bond distances, atomic charges, and electronic properties such as electronegativity and hardness. By encoding these characteristics, the model captures key structural and energetic trends that influence reaction feasibility.
A key advantage of this ML approach is its computational efficiency, enabling rapid screening of chemical spaces to identify feasible molecular structures. AI-driven models trained on reaction energy barriers refine and enhance structure determination processes. Furthermore, continuous integration of experimental and computational data enhances predictive accuracy, ensuring adaptability to evolving trends in molecular chemistry.
Computational screening and quantum chemistry approach
A computational study validated and refined ML-based energy barrier predictions. Advanced quantum chemistry techniques were applied to analyze specific molecular interactions and assess the feasibility of transformations. The results confirmed that while some reactions proceed smoothly, others require additional energy due to structural and electronic differences.
This study illustrates how ML-generated predictions can be verified and improved using quantum chemical methods, leading to a more systematic and reliable approach to molecular structure prediction. By analyzing a wide range of possible reactions, the study helps refine search strategies for viable molecular transformations and enhances AI-driven structure determination. Due to the high accuracy achieved with the ML model, expensive computations can be eliminated.
Recovering important protein configurations with coarse-grain models
Predicting stable protein structures plays a pivotal role in drug discovery and therapeutic advancements. Traditionally, molecular dynamics simulations are used to explore, discover, and validate configurations, but for large proteins, this process is computationally expensive. Coarse-grain models simplify these simulations but often lack chemical accuracy or detect only limited stable states.
By using ML to replace traditional simulations with faster but still accurate coarse-grain models, these problems have recently been circumvented. ML coarse-grain models reduce complex simulations to a few representative atoms while preserving structural variety. Recent ML-driven coarse-grain models not only enhance molecular configuration prediction across diverse proteins but also establish a pathway for versatile, chemically accurate, and efficient structure prediction models.
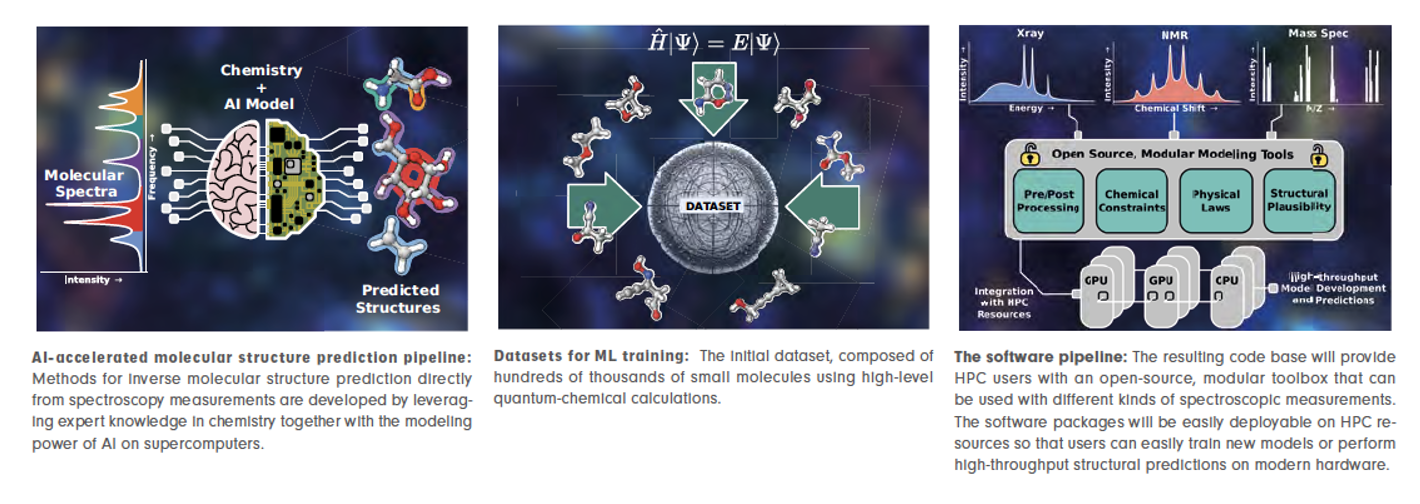
AI-accelerated molecular structure prediction pipeline on supercomputers
Building on prior work, we advance AI-based molecular structure prediction. Our methodological design and implementation target state-of-the-art supercomputer resources at NHR@ZIB, combining computationally demanding molecular simulations with AI/ML-based predictive modeling to analyze patterns and make predictions from large datasets.
We are currently developing a structure prediction method for molecules based on computed spectra, advancing the direct prediction of molecular structures from infrared (IR) spectroscopy data – a crucial frontier in molecular science. Since IR spectra are challenging to analyze manually, we train AI models on quantum chemistry datasets to enable highly accurate predictions.
Outlook
Computational chemistry, ML, and supercomputers provide a robust framework for structure prediction. The ML approach accelerates energy barrier estimation and broadens the scope of possible molecular transformations, while quantum chemical studies validate and refine these predictions. By integrating these methodologies with experimental data analysis, particularly IR spectroscopy, we are developing state-of-the-art AI-driven models capable of accurately predicting molecular structures. This approach advances molecular science with broad scientific and industrial applications.
