Die Modelle in den Lebenswissenschaften werden immer genauer, was ihre Annäherung an die Realität angeht. Parallel dazu werden neue Technologien und Techniken für die Berechnung solch großer und datenintensiver Probleme auf Hochleistungsrechnern entwickelt. In unserer Gruppe überbrücken wir diese Lücke, indem wir biowissenschaftliche Modelle entwerfen, die auf Hochleistungsrechnern effizient berechnet werden können. Dieser Schwerpunkt ermöglicht es uns, realistische Modelle zu berechnen, die dann in anderen lebenswissenschaftlichen Disziplinen verwendet werden können.
Ein Forschungsgebiet unserer Gruppe ist die Entwicklung neuer Medikamente, die in der Regel zeitaufwendig ist und teure sogenannte "wet lab" Versuche erfordert. Das strukturbasierte virtuelle Screening hat das Potenzial, dies zu beschleunigen, indem potenzielle Arzneimittelkandidaten (Verbindungen) virtuell an einen Rezeptor angepasst werden. Mit Hilfe hierarchischer Strategien, KI-basierter Ansätze und mathematischer Methoden wollen wir das virtuelle Screening hocheffizient und anwendungsnah gestalten.
Konkret arbeiten wir an Erweiterungen der Open-Source-Screening-Plattform Virtual Flow, die perfekt skaliert und eine frei verfügbare Bibliothek mit mehr als 1,4 Milliarden Molekülen bietet.
Drug Candidates as Pareto Optima in Chemical Space | |
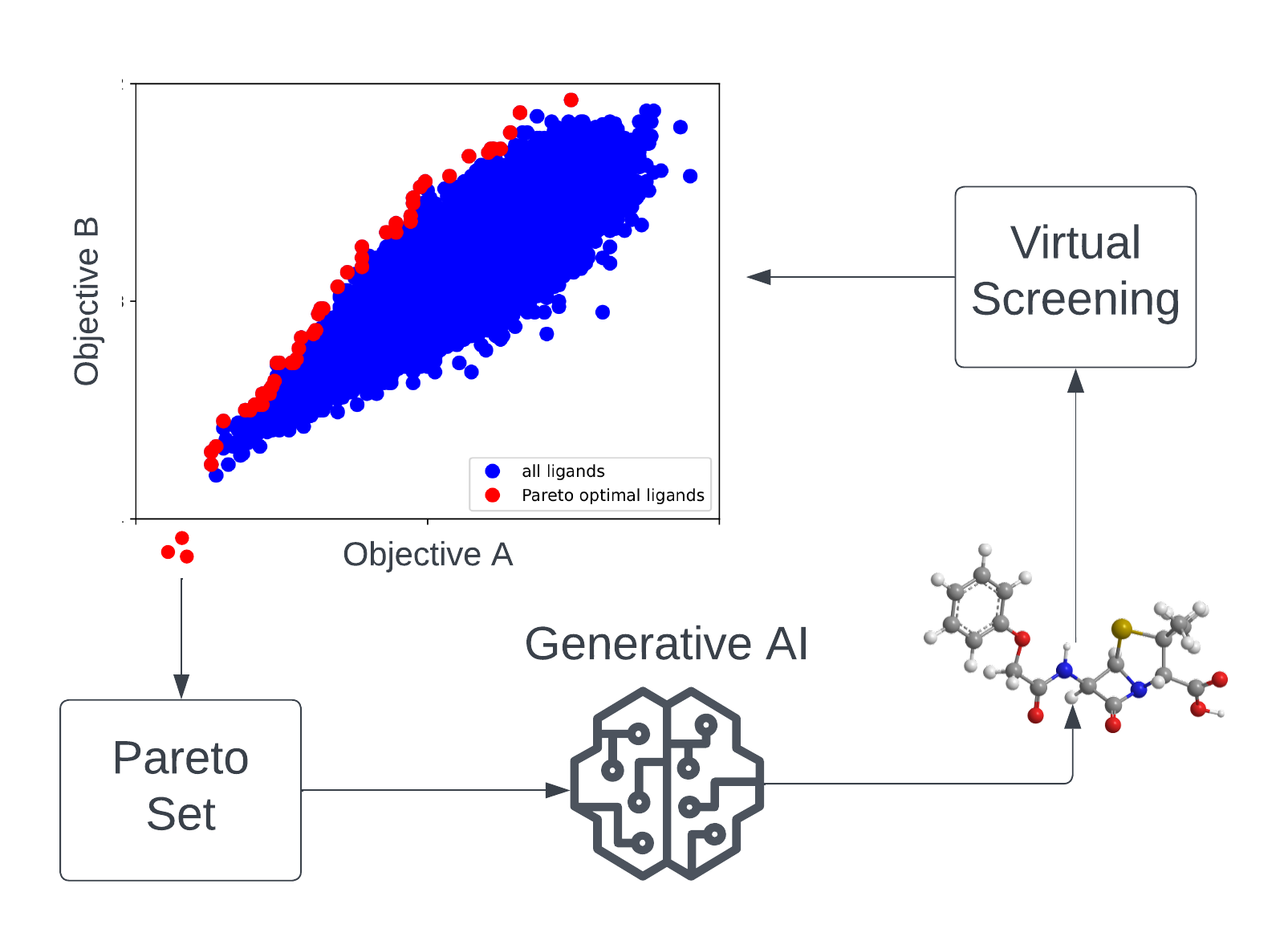 | Die Suche nach neuartigen Medikamentenkandidaten, die gleichzeitig hochwirksam sind, definierte chemische Eigenschaften erfüllen und zudem geringe Off-Target-Effekte zeigen, kann als Multi-Objective-Optimierungsproblem (MOOP) im chemischen Raum angesehen werden. Im MATH+-Projekt „AA1-19 Drug Candidates as Pareto Optima in Chemical Space“ werden KI-basierte generative Modelle verwendet, um neuartige wirkstoffähnliche Moleküle aus dem chemischen Raum über bestehende Ligandenbibliotheken hinaus für therapeutische Ziele mit ungedecktem medizinischem Bedarf zu identifizieren. Aufgrund des extrem großen Suchraums (ca. 10⁶⁰ wirkstoffähnliche Moleküle) werden in diesem Projekt effiziente Lernalgorithmen und virtuelle Hochdurchsatz-Screening-Methoden entwickelt und angewendet. |
High-performance computing and machine learning for drug discovery | |

| Um komplexe Systeme zu simulieren, die es ermöglichen, das Verhalten von Molekülen unter realen Bedingungen zu untersuchen, sind Hochleistungsrechner erforderlich. Dieses Projekt kombiniert künstliche Intelligenz und parallele Algorithmen, um Moleküle auf einem Supercomputer präziser und effizienter zu berechnen. Mit Hilfe des am ZIB entwickelten IsoKANN, können für die Wirkstoffentwicklung wichtige Übergangsraten effizient berechnen werden.
|
An open-source platform for ultra-large virtual screening: VirtualFlow | |
VirtualFlow ist eine Open-Source-Workflow-Plattform zur Durchführung extrem großer virtueller Screenings auf Hochleistungs-Computing-Clustern oder in der Cloud (https://github.com/VirtualFlow). Die Plattform lässt sich perfekt skalieren und ermöglicht dadurch das Screening von bis zu einer Milliarden kleiner Moleküle auf ein bestimmtes Ziel. Unsere Gruppe entwickelt die Plattform aktiv mit. Weitere Informationen zum VirtualFlow-Projekt finden Sie unter https://scholar.harvard.edu/gorgulla/virtualflow-project
| |
