Binding of a multivalent ligand to a multivalent receptor consists of kinetically different elemetary steps. The first binding event is an intermolecular process, whereas, the subsequent steps are intramolecular. Thus, understanding the timescales of these different steps and the characterization of the "rebinding" effect is a mathematical challenge.
In this project we develope methods for the multiscale simulation and the extraction of kinetical information from multivalent molecular binding processes.
"Multivalency as chemical organisation and action principle: New architectures, functions and applications". Subproject C2 aims at determining entropy and enthalpy differences between different multivalently binding systems, using the conformation dynamics approach. Qualitative and quantitative results of these calculations are intended to enhance the understanding of multivalency in chemical systems. Finally, these findings are hoped to contribute to designing optimal molecular spacers for practical applications of multivalent binding. In this context, central research topics include the development of efficient and stable numerical methods for high-dimensional conformation spaces, as well as the mathematical modelling of energy contributions involved in multivalent binding.
Preliminary work
As preparatory work for the sucessful DFG examination in 2007, we dealt with protein-resistant surfaces. Simulation experiments helped to link the effectiveness of protein resistance to the flexibility of the molecular structures under observation, i.e. entropy differences between the different polymer variants attached to the surface.
Methods, algorithms and software
Using the algorithmic framework of conformation dynamics, advanced sampling methods have been developed to facilitate the computation of equilibrium densities in metastable dynamical systems (Kube, Weber 2008), available in the software package ZIBgridfree. Furthermore, the conformation dynamics approach has been adapted so as to resolve the statistics of chemical binding processes (Bujotzek, Weber 2009). In order to cope with the large-scale chemical systems under observation in Collaborative Research Center 765, there is an ongoing implementation effort to adapt these methods to highly parallel computer architectures. The focus of this effort is to produce efficient and reliable interfaces to well-established high-performance MD software packages such as Gromacs.
Another central question in chemical multivalency is the role of entropy throughout the binding process. Methods have been developed to estimate entropy differences directly from simulation trajectories (Weber, Andrae 2010). Currently, new approaches for statistical reweighting of partial densities gained from decoupled samplings (as, e.g., produced by ZIBgridfree) are under development.
Applications
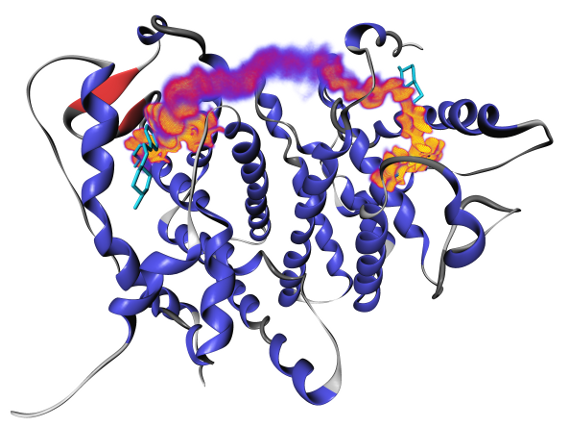
Bivalent estrogen receptor ligands
![]()
Bivalent ligands for human estrogen receptor (ER) represent a promising application of chemical multivalency. In this context, we are working on designing chain-like molecular spacers that are able to bridge the distance between the binding sites with good fit. To achieve this aim, structure, energy and entropy have to be considered. We are also investigating interactions of the spacer with its ligand groups, possibly hindering high-affinity binding. Furthermore, we conduct docking experiments to evaluate novel ER ligands developed by cooperation partners.
Nucleid acid conjugates as molecular spacers
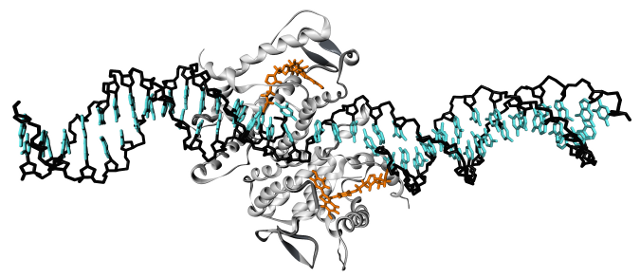
In contrast to the chain-like spacers of the ER project, DNA spacers offer a more defined structure. In this case, however, molecular simulations can help to resove optimal distances between ligand presentations sites and the structural properties of the linker itself. Finally, we want to introduce single-stranded segments into the DNA to determine the implications on the overall flexibility, possibly in context with the dimeric target protein domein Syk-tSH2.
Conformation dynamics of tetraarylporphyrins
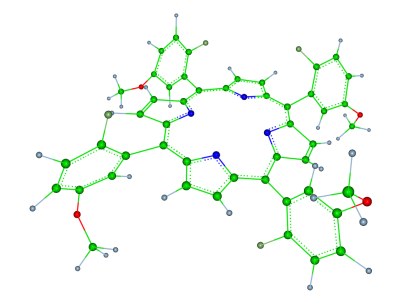
Tetraarylporphyrins are an interesting model for the role of entropy during conformational changes. We are exploring the conformational properties of the tetraarylporphyrine core structure with different substituents of increasing size and molecular weight.
Funding period 2: Multivalent kinetics
By now, Collaborative Research Center 765 has started off into the second funding period. The focus for subproject C2 is the unraveling of multivalent binding processes and the development of new theoretical approaches.
Quantifying the rebinding effect in multivalent ligand-receptor systems
An additional factor that has been proposed to contribute to the multivalency effect is “rebinding”: As soon as a single ligand-receptor complex dissociates, the presence of another ligand “on coat-tails” will increase the probability of another binding event, which in turn will drive the system to a state where all ligands are bound. This phenomenon has also been described in terms of “a locally increased concentration of ligands”, such that ligands which are close to the receptor contribute with higher binding rates to the overall binding kinetics.
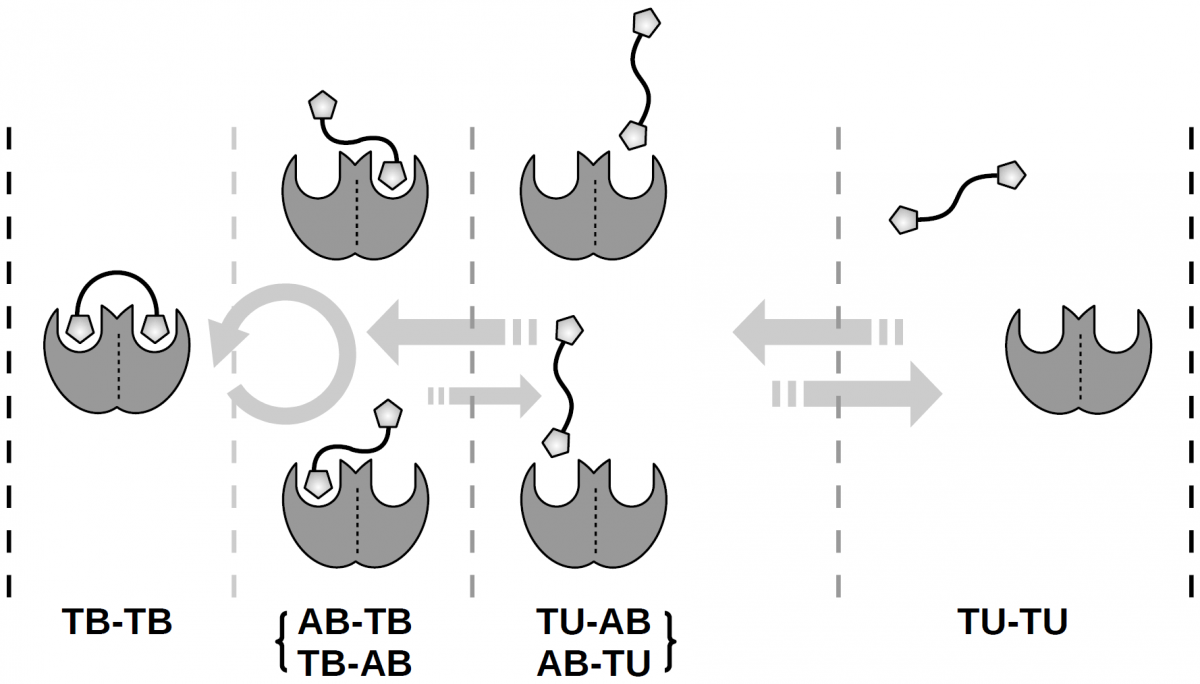
We present a novel approach for the quantitative description of the rebinding effect. In order to model the inherent memory effect of a spacer-connected system, we derive a mathematical approach based on Markov State Models and Conformation Dynamics. These theoretical frameworks are used for extracting the long time dynamics of a given molecular system from (rather short term) simulation trajectories. Both approaches divide conformational space into long-lived (metastable) states, which is equivalent to coarse-graining time. The theoretical investigations are illustrated by studying different prototypic, but highly simplified, ligand-receptor systems regarding the contribution of the rebinding effect. We propose a criterion to determine if and in how far rebinding is going to occur for a given system.
Outlook
Currently, work is directed on tracing rebinding effects in "real-world" ligand-receptor systems and the validation of our model with data from wet chemistry. This is mainly done using the new implementation of ZIBgridfree, which includes the explicit modeling of solvent. Furthermore, new polyvalent polymer-based systems from Collaborative Research Center 765 will be in the focus of our research.