Models in the life sciences are becoming increasingly accurate in terms of their approximation to reality. In parallel, new technologies and techniques for computing such large and data-intensive problems on high-performance computers are also developing. In our group we bridge this gap by designing life science models that can be efficiently computed on high performance computers. This focus enables us to compute realistic models that can then be used in other life science disciplines.
One research field of our group is the development of new drugs, which is typically time consuming and expensive wet lab experiments are necessary. Structure-based virtual screening has the potential to accelerate this by virtually fitting potential drug candidates (compounds) into a receptor. This process requires a high computational effort and asks for smart parallel strategies on computer clusters. Using hierarchical strategies, AI-based approaches, and mathematical methods, we aim to make virtual screening highly efficient and close to application. Specifically, we are working on extensions to the open source screening platform VirtualFlow, which scales perfectly on supercomputer clusters and cloud platforms. It currently provides a freely available library of more than 1.4 billion molecules for virtual screening.
Drug Candidates as Pareto Optima in Chemical Space | |
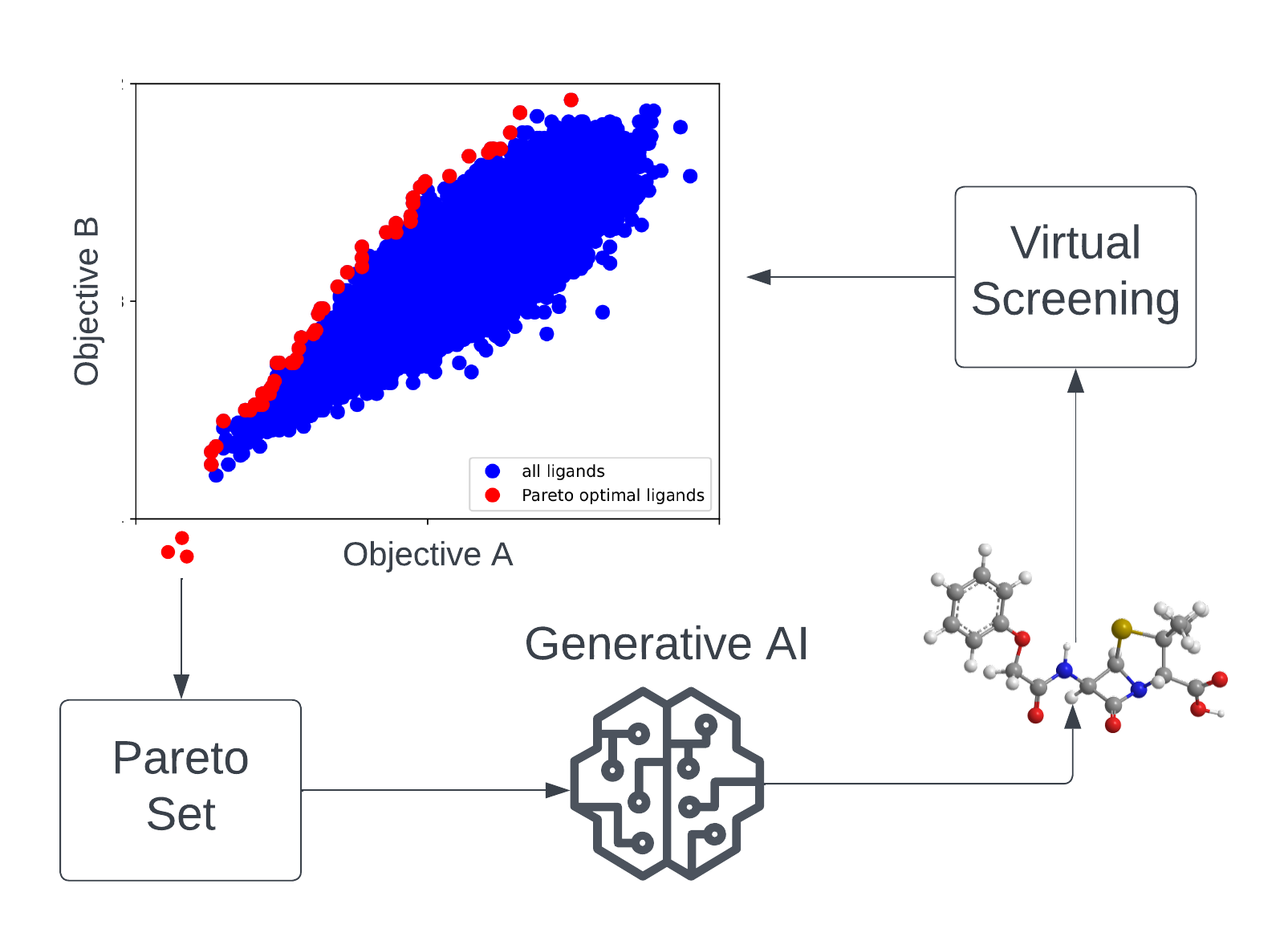 | The search for novel drug candidates that, at the same time, act with high efficacy, comply with defined chemical properties, and also show low off-target effects can be perceived as a multi-objective optimization problem (MOOP) in chemical space. In the MATH+ Project "AA1-19 Drug Candidates as Pareto Optima in Chemical Space" AI-based generative models are used to identify novel drug-like molecules from chemical space beyond existing ligand libraries for therapeutic targets of unmet medical need. Due to the extremely large search space (approximately 10⁶⁰ drug-like molecules), efficient learning algorithms and high-throughput virtual screening methods are developed and applied in this project. |
High-performance computing and machine learning for drug discovery | |

| High-performance computers are required to simulate complex systems that allow to study the behavior of molecules under real conditions. This project combines artificial intelligence and parallel algorithms to calculate molecules more precisely and efficiently on a supercomputer. We are aiming to implement and develop novel computational approaches to facilitate drug discovery and optimization. |
An open-source platform for ultra-large virtual screening: VirtualFlow | |
| VirtualFlow is an open-source workflow engine designed to perform ultra-large virtual screenings on high-performance computing clusters or in the cloud (https://github.com/VirtualFlow). The platform scales perfectly and thereby enables to screen up to billions of small molecules to a given targets. Our group is actively co-developing the platform. More information about the VirtualFlow Project can be found on https://scholar.harvard.edu/gorgulla/virtualflow-project | |
